from the end of the shutdown,-and I have huge hope that we the people will continue to stand together to help each other through the days!-we do get the Astronomy Photo of the Day again!
Explanation: Few cosmic vistas can excite the imagination like The Great Nebula in Orion. Visible as a faint, bland celestial smudge to the naked-eye, the nearest large star-forming region sprawls across this sharp colorful telescopic image. Designated M42 in the Messier Catalog, the Orion Nebula’s glowing gas and dust surrounds hot, young stars. About 40 light-years across, M42 is at the edge of an immense interstellar molecular cloud only 1,500 light-years away that lies within the same spiral arm of our Milky Way galaxy as the Sun. Including dusty bluish reflection nebula NGC 1977, also known as the Running Man nebula at left in the frame, the natal nebulae represent only a small fraction of our galactic neighborhood’s wealth of star-forming material. Within the well-studied stellar nursery, astronomers have also identified what appear to be numerous infant solar systems.
For some people, gender shifts over time, often through changes in one’s sense of self. A transgender man may realize they are nonbinary and stop hormone replacement therapy. A trans woman may face so much discrimination that she represses her identity. And some trans people medically reverse their transition to live as their sex assigned at birth.
These experiences are all part of a process known as detransitioning. Although detransitioning does not have a consistent social or academic definition, it generally applies to someone who has sought a gender transition and then stopped, shifted or reversed aspects of it. Their experiences offer a deeper look at how discrimination and gender norms impact our lives, how gender-affirming care can be improved, and how identity is perhaps more fluid than previously thought.
As experts work to understand detransitioners, their vulnerabilities and their highly individualized needs, their identities are being co-opted as part of a national campaign against transgender rights. Health care access and research are being blocked by politicians for both trans people and detransitioners — while anti-trans rhetoric puts everyone at risk.
The Federal Trade Commission and the Justice Department are investigating gender-affirming care as medical fraud, and they are rooting this effort in detransitioners’ stories that fit the narrative the Trump administration wants to advance. The White House wants the National Institutes of Health to study “regret” and “detransition,” even as it cuts any federal funding for researchthat mentions the word “trans.” The U.S. Department of Education hosted a “Detrans Awareness Day” event last March. Meanwhile, its functions have been severely undermined by layoffs and budget cuts.
The White House and agencies like the Justice Department claim that gender-affirming care is mutilating children, overlooking that young trans people live happily after transition and the studies showing that adolescents who regret transition are in the minority. Government officials describe trans people and detransitioners as victims of a medical conspiracy to boost profits and force gender ideology on families. Now, they are seeking evidence to prove those claims by subpoenaing hospitals for patients’ private data, including doctors’ notes, patient addresses and Social Security numbers.
Gender-affirming care has been broadly endorsed by the medical community for its effectiveness in treating gender dysphoria, a persistent distress felt when one’s body is out of sync with their identity. The 2022 U.S. Trans Survey, which polled over 92,000 trans and nonbinary people 16 and older, found that social and medical transition were profound sources of life satisfaction. Experts and advocates agree that more research and more understanding are needed to improve trans medical care. But under Trump, they also expect transgender and intersex health to keep getting worse, not better.
The 19th spoke with two detransitioners who feel harmed and usedby the Trump administration, which has positioned itself as a protector of those who detransition. Adriana lives in New York City, where she feels safe to express herself among so many LGBTQ+ people, but has struggled to access adequate health care. Ara lives in North Carolina, a state that has several laws restricting trans rights and health care access — and where support from a mental health program and her partner has helped her navigate the challenges of detransitioning. As politicians stoke fear about gender non-conformity, their experiences offer a deeper understanding of what it means to live authentically in a politically volatile time.
Still, more young people have been exploring their identities, expanding the boundaries of gender and adding to the cultural and social norms surrounding it. Detransitioners’ experiences are part of that social evolution. Their stories of regret and pain exist alongside stories of joy and empowerment — and these are all part of a journey of self-discovery that may have turned out to be more complicated than they initially thought. The question is, will elected officials support them on this journey or cause more harm?
‘Taking away trans health care is taking away people’s lives’
As providers of trans health care have become political targets, Ara Kareis’ own routine treatments have been disrupted.
‘I just feel like such a power source’
The joy that Adriana Del Orden feels in her body could have only come through exploring her gender. She’s tired of being told that she ruined her life.
The politics of detransition
Political rhetoric doesn’t capture the complexity of detransitioning — or what taking away health care means.
The shutdown may finally be coming to an end. Our health care costs, unfortunately, will not. I’ll be back on Wednesday with a deep dive into just how spectacularly absurd our health care system has become.
In the meantime, RSV and flu are picking up speed, and a concerning infant formula recall has been linked to a rare botulism cluster, thanks to a small but mighty team in California. The FDA is expected to remove the black box warning from hormone replacement therapy—a move that’s scientifically sound but bound to spark drama from HHS. We also have new blood pressure guidelines that could make prevention a lot more personal. And amidst it all, a few more pieces of genuinely good news to end on a high note.
Let’s dive in!
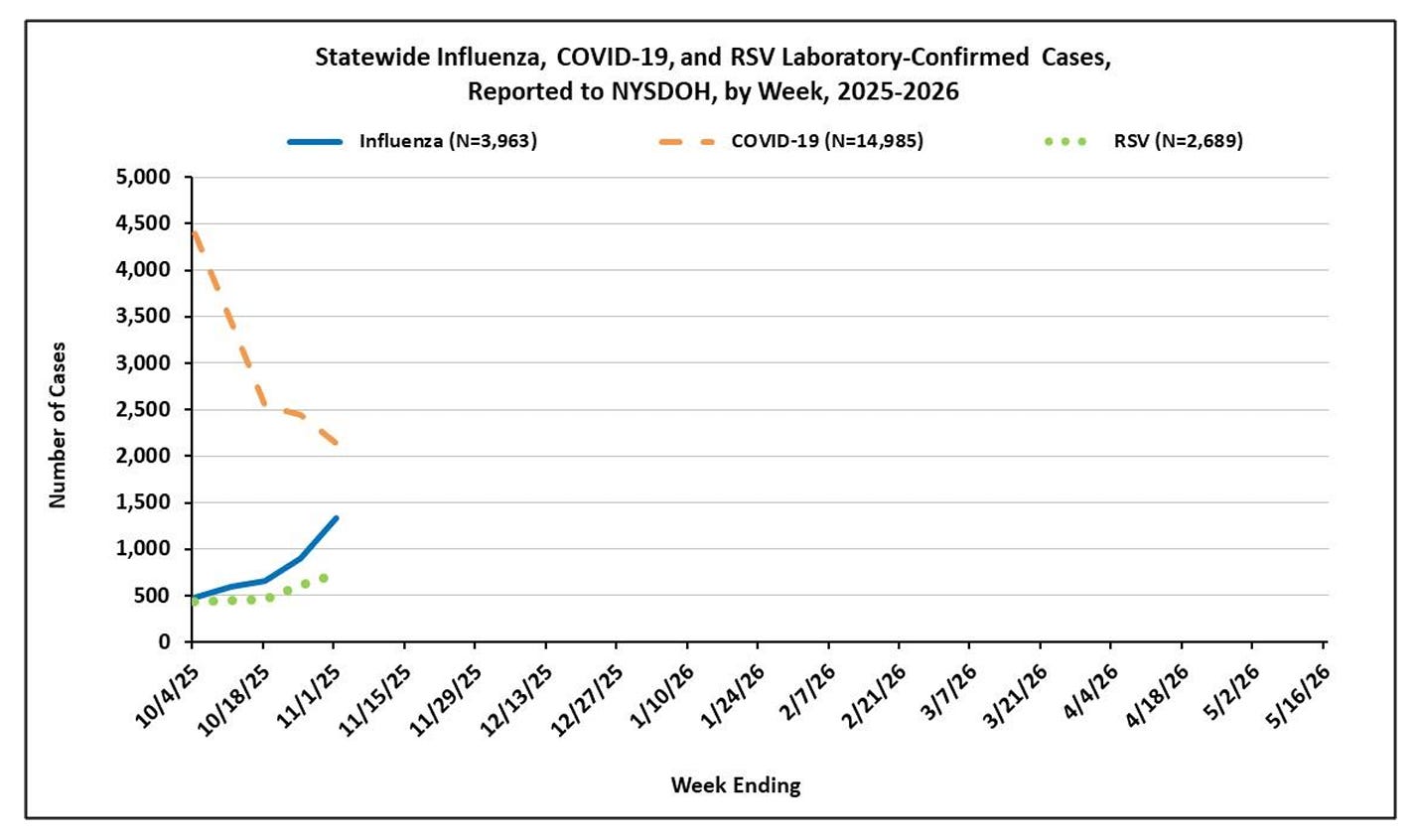
Disease “weather” report: RSV and flu gaining momentum
It will take some time for the CDC data systems to ramp up again after being offline for over 40 days. For now, we’ll continue to rely on the alternative sources, such as Dr. Caitlin Rivers’ updates and the PopHive dashboards.
RSV continues to climb slowly but steadily, especially among children under five. National growth is still linear—not yet exponential—but that acceleration could occur at any time.
Theflu remains relatively low but is beginning to increase, particularly among young children. As Dr. Rivers notes, “Hawaii has moved to moderate activity, Arizona has surpassed its seasonal baseline, and New York cases jumped 49% in the past week.”
Source: New York State Department of Health Respiratory Surveillance Report
U.S. childhood flu vaccination rates have dropped from 62% to 49% over the past five years. Last year saw one of the deadliest seasons on record, with 280 pediatric deaths—the highest since tracking began in 2004. About 90% of those children weren’t fully vaccinated. Our deadliest flu season came at a time of historically low vaccination rates, which can’t be a coincidence. We don’t yet know this season’s coverage, but if it falls further, we could be facing another tragic record.
Covid-19 remains in a lull, though we typically see a winter rise starting in mid-to-late November.
I’m really hoping these three viruses don’t peak simultaneously. Hospitals strain under just a bad flu season; I couldn’t imagine the “big three” all at the same time. Historically, their peaks have staggered, but given how little we truly understand about these overlapping patterns, that may have been more a matter of luck than a rule. Time will tell.
What this means for you: This is the best time to get vaccinated. It’s certainly not too late.
Infant formula outbreak and Listeria in pasta
Over the weekend, a troubling cluster of infant botulism cases was linked to ByHeart Whole Nutrition Infant Formula. Even during a government shutdown and an increasingly challenging environment, outbreak teams have been working around the clock to protect our most vulnerable.
What this is: Infant botulism is extremely rare but serious. It occurs when Clostridium botulinum spores—commonly found in soil, dust, and some foods—germinate in a baby’s intestines and release toxins that can paralyze muscles, interfere with breathing, and require intensive care. In a typical year, the U.S. sees 160–180 cases, often linked to environmental exposure or foods like honey. Even a small cluster of cases is a clear red flag.
What we know:Clostridium botulinum spores have been detected in ByHeart infant formula, resulting in the hospitalization of 13 infants across 10 states. The California Department of Public Health (CDPH) played a key role in identifying this cluster. CDPH is the only source in the world for BabyBIG—the lifesaving antitoxin—and manages all clinician calls and treatment distribution. Their team noticed a spike in requests and discovered that the affected infants all consumed the same formula brand, prompting an alert to CDC. Importantly, ByHeart produces just 1% of U.S. infant formula, so this alone is unlikely to cause a national shortage. (Be sure to sign up for YLE CA for a deeper dive this Thursday.)
What we don’t know: Epidemiologists are investigating whether contamination is truly confined to ByHeart or reflects a broader issue in the manufacturing or ingredient sourcing process.
What to do:Stop using ByHeart Whole Nutrition Infant Formula immediately. Retailers should pull it from shelves, including Amazon, Kroger, Walmart, Whole Foods, Target, and Sam’s Club.
There is also the ongoing Listeria outbreak associated with frozen pasta dishes, including some sold at Trader Joe’s and other grocery chains. We’ve covered this before, but according to the agency’s ongoing investigation, two more brands of food are linked to the outbreak. There’s a long list of recalled products you can find here. Throw out immediately.
In total, there have been 27 illnesses reported, 25 hospitalizations, and six deaths in 18 states since late September.
Number of cases associated with the pasta Listeria outbreak. Source: CDC
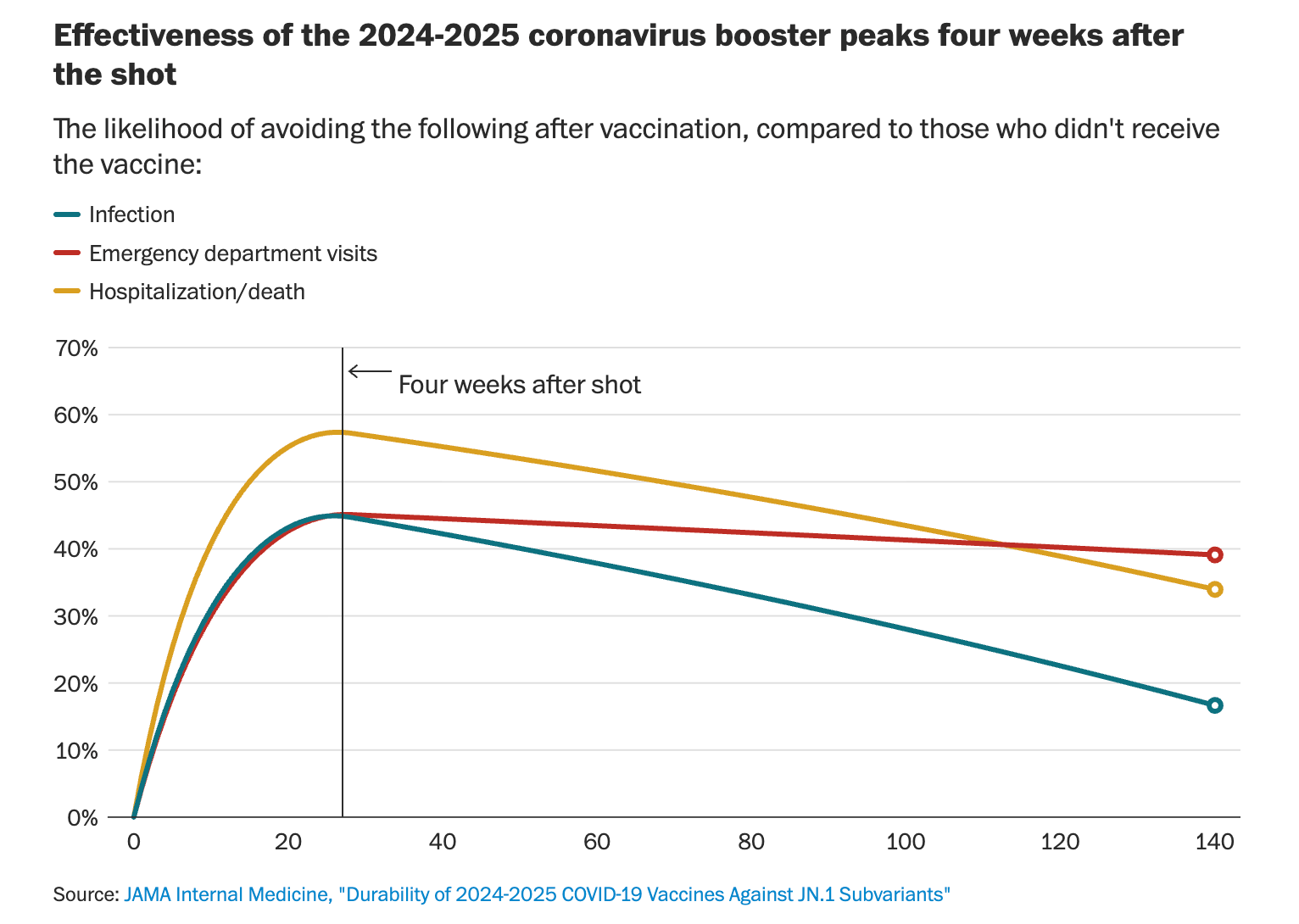
HRT ‘black box’ warning may be gone soon
Word is that the FDA will remove the black box warning from hormone replacement therapy (HRT) this week. (It may now be pushed back with the government reopening.) If this announcement reflects the HRT review conducted at HHS last month, it will likely be messy and riddled with inaccuracies. However, if you look at the science without the drama, removing the black label is not unreasonable.
The black box warning—the strictest warning label, meant for drugs with potentially life-threatening risks—was enacted after a 2002 Women’s Health Initiative study found increased risks of stroke and breast cancer. Since then, research has revealed crucial nuance: risks depend on timing, dose, and delivery. Starting HRT earlier, using low-dose or localized estrogen, and tailoring therapy to each woman can be both effective and safe.
What this story is really about: Menopause is universal, yet too often misunderstood. While the science continues to evolve, the major problem is that clinical practice and training have lagged far behind. Women deserve evidence-based care, informed clinicians, and the freedom to make choices rooted in both science and compassion. We are still far from what is needed for women.
POLL
(snip-poll won’t embed here; go to the Substack page. The question is if readers would like to see a deep dive into HRT. If you wish to vote, click “read on Substack”, above.)
New blood pressure guidelines
Major new blood pressure guidelines dropped for the first time in years.
At the center of the update is an enhanced assessment tool called the PREVENT calculator, developed by the American Heart Association. It’s designed to estimate a person’s 10-year risk of cardiovascular disease using factors like blood pressure, cholesterol, smoking, and diabetes. The model was built using data from 6.5 million U.S. adults aged 30 to 79, making it one of the most representative tools available.
Here’s what stands out:
A new threshold for medication. The key number to watch is 7.5%. If your 10-year risk of heart disease is at or above that level, physicians are now encouraged to consider medication even if your blood pressure hovers around 130/80 mmHg. If your risk is below that, lifestyle changes, like healthy eating, exercising, and better stress management, remain the first step.
A focus on home monitoring.The guidelines also emphasize checking blood pressure at home. Growing evidence shows that home readings may actually be more accurate predictors of long-term risk than in-office measurements. So spending $45 on an arm monitor if you have hypertension might be a great (even lifesaving!) idea.
What this means for you: This makes heart health more personalized than ever. You can calculate your own 10-year risk using the PREVENT calculator (although you will need some pretty specific numbers from your last blood panel). Regardless, aim for blood pressure readings below 130/80, and use these new tools to guide smarter—and earlier—prevention.
Good news!
Here are some of the great things worth highlighting:
The American Academy of Pediatrics (AAP) is standing up for science. The organization filed an updated lawsuit last week against HHS Secretary Robert F. Kennedy, Jr., asking a court to disband a panel (ACIP) appointed by RFK Jr. and to overturn recent decisions made by that panel. Then, to proceed under court supervision. The legal move is a direct push to restore expert-led vaccine policymaking.
The Vaccine Integrity Project (VIP) is stepping up on Hep B. With the next ACIP meeting coming up in early December, where the agenda will likely include the Hepatitis B vaccine for infants, the childhood vaccination schedule, and HPV—VIP, an independent group of scientists formed in response to waning trust in RFK Jr.’s ACIP, is conducting an evidence review ahead of what could be a contentious meeting. Their work helps ensure the science remains front and center.
Vaping among youth has seen a decline; but we still have a long way to go. A new study has found that the number of U.S. teens vaping has decreased overall. That’s progress. But among those who still do it, vaping is becoming more frequent and harder to quit—signs that use is shifting from experimentation toward dependence for some. If you have a teen who vapes, check out the EX Program, which is a free, anonymous text-messaging program designed specifically for young people who vape. There’s also SmokefreeTXT for Teens.
Question grab bag: You ask, we answer!
How long does the Covid-19 vaccine last? I got mine in September, do I need to get another?
A study published last week confirmed what we’ve consistently seen: protection against Covid-19 wanes over time. A study of more than 1.8 million Americans from the previous season showed protection against infection and severe disease declined after 4-5 months. The findings underscore the benefit of getting a Covid-19 vaccine every six months, especially for adults over 65, who accounted for nearly 80% of hospitalizations in the study.
In case you missed it
Help shape our AI + Health conversation. Thanks to everyone who responded to our survey last week! We’re running the AI and health survey to hear your thoughts on using tools like ChatGPT, Claude, Copilot, and PerplexityAI to get health information. If you missed it, take the survey here.
New York YLE’s Marisa discussed the state reaching a historic low in youth tobacco use.
California YLE Matt discussed the impact of ICE raids on access to healthcare.
WNV linked each of these. Here are the original pages with snippets.
After No Kings, It’s Time to Escalate by Eric Blanc
We need bigger—and more disruptive—nonviolent campaigns that can go viral and peel away Trump’s pillars of support Read on Substack
American democracy is on the ropes. Trump and his billionaire backers are doing everything possible to transform our country into an authoritarian state like Hungary or Russia, where the trappings of institutional democracy mask brazen autocratic rule.
Our president’s sinking popularity numbers might not matter so much if his administration is either able to ignore electoral results or to distort the electoral map so badly that there’s almost no way to vote Republicans out.
Far too many Democrats and union leaders naively hoped that the courts would save us. But the Supreme Court has given a green light to Trump’s power grab, and it appears poised to overturn Section 2 of the Voting Rights Act, the last major legal roadblock to prevent Republicans from disenfranchising millions of Democrats and Black voters across the South.
Are we cooked? Trump would certainly like us to believe he’s unstoppable. Faced with the administration’s relentless offensive against immigrants, free speech, public services, and majoritarian rule, it’s normal to sometimes succumb to despair. But there’s no need to throw in the towel — and there are concrete next steps we can all take to win back the country through nonviolent resistance. As Chicago Teachers Union (CTU) president Stacy Davis Gates reminds us, Trumpism “won’t be stopped just in the courts or at the ballot box.” (snip-there is MORE on the page linked at “Read on Substack” above)
=====
The introvert’s guide to fighting for democracy by Protect Democracy
Six ways to protect democracy — without attending a protest Read on Substack
If you’re reading this, you’re concerned about our democracy’s slide into authoritarianism — and you want to do something about it. Wahoo! You’ve taken the first and most difficult step: committing to action.
Now come the fun parts.
I want to be really clear on a couple things to start out. First, there is no one-size-fits-all best way to exercise your First Amendment rights of speech and association. Every successful social movement has employed a wide variety of tactics and repeatedly adjusted to respond to facts on the ground. Opt for action over agonizing about optimal tactics.
Second, be realistic. We are all busy. Reflect on the commitments you can actually sustain with room to grow. It is far better to regularly move the ball forward on a smaller effort than to dive into and never complete an ambitious one.
Third, be unique! You have unique talents, skills, and passions. Let those guide your advocacy. Focus on projects that bring you joy, things you actually look forward to engaging with week after week. Lean into the comparative skills and expertise you bring to the movement.
With all that in mind, here’s a short list of six ways everyone can protect democracy — even (especially) if going to a protest or some other more public form of engagement isn’t for you.
1. Check in with your local library
Local libraries are the backbone of an informed democratic citizenry, and they provide crucial resources for underserved communities. But their funding is under attack by the administration, which has cut critical funds nationwide.
So, give the library in your neighborhood a call. See how they are doing in relation to funding cuts and if there are ways you can support them. Do they take book donations? Need volunteers? See if there are teach-in or reading groups you can join — or even lead. Offer to help curate pro-democracy reading lists for various ages. Many libraries are open to suggestions for books to add to the collection — here are some recommendations from our team.
2. Fill the gaps left by government programs
Taking care of one another is essential movement building. Check in on your food pantry and community kitchen — many of which have faced funding cuts — to see how you can help. (snip-MORE at the page linked above: “Read on Substack”)
unless someone’s able to override an override of an override. I’m not familiar with this publication; this was another story I read during a jog, but the story seems feasible. Experts here will know. I’m just tossing it in here.
The smart vacuum cleaner was remotely bricked for not collecting data.
An engineer got curious about how his iLife A11 smart vacuum worked and monitored the network traffic coming from the device. That’s when he noticed it was constantly sending logs and telemetry data to the manufacturer — something he hadn’t consented to. The user, Harishankar, decided to block the telemetry servers’ IP addresses on his network, while keeping the firmware and OTA servers open. While his smart gadget worked for a while, it just refused to turn on soon after. After a lengthy investigation, he discovered that a remote kill command had been issued to his device.
He sent it to the service center multiple times, wherein the technicians would turn it on and see nothing wrong with the vacuum. When they returned it to him, it would work for a few days and then fail to boot again. After several rounds of back-and-forth, the service center probably got tired and just stopped accepting it, saying it was out of warranty. Because of this, he decided to disassemble the thing to determine what killed it and to see if he could get it working again.
Since the A11 was a smart device, it had an AllWinner A33 SoC with a TinaLinux operating system, plus a GD32F103 microcontroller to manage its plethora of sensors, including Lidar, gyroscopes, and encoders. He created PCB connectors and wrote Python scripts to control them with a computer, presumably to test each piece individually and identify what went wrong. From there, he built a Raspberry Pi joystick to manually drive the vacuum, proving that there was nothing wrong with the hardware.
From this, he looked at its software and operating system, and that’s where he discovered the dark truth: his smart vacuum was a security nightmare and a black hole for his personal data. First of all, it’s Android Debug Bridge, which gives him full root access to the vacuum, wasn’t protected by any kind of password or encryption. The manufacturer added a makeshift security protocol by omitting a crucial file, which caused it to disconnect soon after booting, but Harishankar easily bypassed it. He then discovered that it used Google Cartographer to build a live 3D map of his home.
This isn’t unusual, by far. After all, it’s a smart vacuum, and it needs that data to navigate around his home. However, the concerning thing is that it was sending off all this data to the manufacturer’s server. It makes sense for the device to send this data to the manufacturer, as its onboard SoC is nowhere near powerful enough to process all that data. However, it seems that iLife did not clear this with its customers. Furthermore, the engineer made one disturbing discovery — deep in the logs of his non-functioning smart vacuum, he found a command with a timestamp that matched exactly the time the gadget stopped working. This was clearly a kill command, and after he reversed it and rebooted the appliance, it roared back to life.
So, why did the A11 work at the service center but refuse to run in his home? The technicians would reset the firmware on the smart vacuum, thus removing the kill code, and then connect it to an open network, making it run normally. But once it connected again to the network that had its telemetry servers blocked, it was bricked remotely because it couldn’t communicate with the manufacturer’s servers. Since he blocked the appliance’s data collection capabilities, its maker decided to just kill it altogether. “Someone—or something—had remotely issued a kill command,” says Harishankar. “Whether it was intentional punishment or automated enforcement of ‘compliance,’ the result was the same: a consumer device had turned on its owner.”
Unfortunately, many other smart vacuum brands use similar hardware, so it’s not far-fetched to think that they have the same setup. This is likely especially true for cheaper devices that have less capable hardware and aren’t capable of edge computing, meaning they’ll have to send the data to some faraway server for processing. But because your information is being offboarded to another device outside of your control, you really have no idea what’s happening to it, giving the manufacturer free rein to use it as it pleases.
In the end, the owner was able to run his vacuum fully locally without manufacturer control after all the tweaks he made. This helped him retake control of his data and make use of his $300 software-bricked smart device on his own terms. As for the rest of us who don’t have the technical knowledge and time to follow his accomplishments, his advice is to “Never use your primary WiFi network for IoT devices” and to “Treat them as strangers in your home.” (snip)
Cosmos Magazine has changed its online presence, while still providing the informational and neat articles they’re known for. Here are a couple for this week.
A near complete fossil rhinoceros has been found on an Arctic Canadian island, making it the most northerly rhino species ever.
Epiatheracerium itjilik [eet-jee-look] is described for the first time in a paper published in the journal Nature Ecology & Evolution. The “Arctic rhino” lived about 23 million years ago during the early Miocene epoch.
The new species was found in fossil-rich lake deposits in Haughton Crater on Devon Island which is part of the northern-most Canadian territory of Nunavut. Devon Island lies at a latitude of about 75°N, well within the Arctic Circle. It is also the largest uninhabited island in the world. (snip-MORE)
The largest low-frequency radio image of the Milky Way ever assembled has captured an unprecedented view of the galaxy, enabling astronomers to study the life stages of stars in new ways.
The data was captured by the Murchison Widefield Array (MWA) radio telescope at Inyarrimanha Ilgari Bundara, the CSIRO Murchison Radio-astronomy Observatory on Wajarri Yamaji Country in Western Australia.
“This low-frequency image allows us to unveil large astrophysical structures in our Galaxy that are difficult to image at higher frequencies,” says Associate Professor Natasha Hurley-Walker from the International Centre of Radio Astronomy Research (ICRAR). “No low-frequency radio image of the entire Southern Galactic Plane has been published before, making this an exciting milestone in astronomy.”
Hurley-Walker is principal investigator of one of the extensive surveys used to construct the image, the GaLactic and Extragalactic All-sky MWA (GLEAM) survey. (snip-MORE, and it’s interesting)
Protesters in support of LGBTQ+ rights and against book bans demonstrate outside of the U.S. Supreme Court Building while the justices heard arguments for the case of Mahmoud v. Taylor in Washington, DC., April 2025
Opinion: In Mahmoud v. Taylor, the justices gave bigotry a permission slip and ruled that parents can “opt out” of LGBTQ-inclusive lessons, further diminishing lessons and practices on inclusivity in civic society, argues Darek M. Ciszek.
The U.S. Supreme Court made a decision earlier this summer that has a significant impact on classrooms nationwide. In their 6-3 decision in Mahmoud v. Taylor, the majority completely missed the point as to why LGBTQ-inclusive education matters. By giving parents the option to pull their kids out of lessons that include LGBTQ+ characters or content, the Court prioritized personal religious objections over creating schools where students can learn without feeling invisible.
Justice Alito‘s majority opinion is especially troubling. He treats LGBTQ-inclusive education as if it were some optional “add-on” that schools can easily work around. As a former teacher, I can confidently say that is not how education works, especially when it comes to curriculum and lesson planning. And while Justice Thomas calls LGBTQ-inclusive education “ideological conformity,” he fails to see that most LGBTQ+ adults today grew up in a school system that forced us to conform to a cisgender and straight worldview. Ironically, I’d consider the Court’s narrow view of public education to be ideologically driven.
Let’s be clear about what LGBTQ-inclusive education is and isn’t. When teachers include books like Uncle Bobby’s Wedding in their curriculum, they are not trying to convert anyone’s child or attack anyone’s faith. They are trying to show students that families come in all colors, shapes, and sizes, reflecting our diverse society.
LGBTQ+ people are also part of every community. We have always been a part of human history, and we deserve to be represented in our nation’s schools. The goal is not to change what students believe at home; it is to teach them how to be respectful in a democratic and diverse world. Luckily, in her dissent, Justice Sotomayor got it right when she said that LGBTQ-inclusive education is “designed to foster mutual civility and respect.”
I could not agree more.
But here’s what the Court’s majority really got wrong: they ignored the anti-bullying efforts that motivate many LGBTQ+ inclusive education programs in the first place. According to the latest National School Climate Survey from GLSEN, 68% of American students reported feeling unsafe in school due to their SOGIE (sexual orientation, gender identity, and/or gender expression) characteristics.
That is two out of three LGBTQ+ youth.
These aren’t just statistics. These are real children trying to learn while dealing with a school environment that tells them, whether implicitly or explicitly, that their identities or families are somehow wrong or shameful.
When schools include diverse families in their lessons, they are not pushing an agenda. They are teaching kids that being different does not mean bad. They are giving LGBTQ+ students a chance to see themselves reflected in their education and helping other students see and understand those who are different from them.
Research shows inclusive education works. Studies have found that an LGBTQ-inclusive curriculum can improve the social and emotional well-being of LGBTQ+ youth. When kids learn about different types of families early on, they are more likely to treat their classmates with kindness instead of cruelty. In other words, when implemented correctly, LGBTQ-inclusive education can be an essential anti-bullying and student well-being strategy.
For instance, as a result of my doctoral research, I have learned that some schools around the world are starting to address LGBTQ+ bullying head-on, and, not surprisingly, it’s through curriculum and instruction. In Scotland, LGBTQ-inclusive education became required in 2021 across both primary and secondary, and most major subject areas. When I interviewed government staff about their experience implementing the new policy, I learned that they even worked with religious groups to inform the effort. Faith communities could agree that inclusion was important for reducing homophobic bullying, even if they had some religious concerns. Scottish students now learn how homophobic language hurts people and develop the social-emotional skills needed for creating safer schools. It’s not ideological instruction; it’s teaching kids critical peer relationship skills.
Similar to the Scottish experience, the U.S. Supreme Court could have left the door open for education authorities to find a balance that respects both religious families and vulnerable LGBTQ+ kids. Real inclusion programs do not ask anyone to abandon their faith. They ask people to treat others with respect and dignity, a lesson I believe everyone should support in class. Kids can learn that some families have two moms without being told their family is wrong. They can remember that using “gay” as an insult hurts people without abandoning their religious beliefs. Getting to know your neighbor does not go against faith.
Unfortunately for the U.S., the impact of the Court’s decision may be severe and widespread, especially in ideologically conservative states. Instead of dealing with complicated opt-out policies, I fear many school districts will probably remove LGBTQ+ inclusive materials entirely. Unfortunately, it can be easier to bow to political pressures than to fight, especially when faced with potential lawsuits or a loss of school funding. This means LGBTQ+ kids lose representation, and all students miss out on critical lessons in diversity and inclusion.
The Court’s decision also has broader implications beyond the LGBTQ+ community. By way of a new precedent, the case approves a heckler’s veto, allowing parents to claim a religious objection to any educational content they may not align with at home. This is because the majority opinion wasn’t apparent on how opting out of inclusive education would work in practice, or what would even qualify as a personal religious objection. We might start seeing opt-out forms for instruction on topics like human evolution, women’s rights, or civil rights history. Thanks to the Court, there is no line in the sand.
When we remove students from lessons about diverse communities, we fail everyone. But the call for truly inclusive education is not going anywhere. Our kids—all of our kids—deserve better.
Darek M. Ciszek is a PhD Candidate in Education at UCLA with a research focus on curriculum, learning, and social development.
Voices is dedicated to featuring a wide range of inspiring personal stories and impactful opinions from the LGBTQ+ community and its allies. Visit Advocate.com/submit to learn more about submission guidelines. Views expressed in Voices stories are those of the guest writers, columnists, and editors, and do not directly represent the views of The Advocate or our parent company, equalpride.
things that are just wrong about this; things to be said about him being full of BS; things to be said about him being full of himself; that he presents as if he is actually designing and building these; that he names them Optimus (from Optimus Prime, a hero in “Transformers”), and so on, and so on, and so on…
In a Tesla earnings call Wednesday, the world’s richest man pondered the future of his company’s Optimus robots—and his control over them.
Tesla might be an electric auto maker, but CEO Elon Musk has made clear that he thinks of it as much more: an innovator in artificial intelligence and software, a builder of world-shaking robots. He’s also argued that Tesla should be worth a lot more than it is today: up to $20 trillion, he posted in July, more than five times the current worth of Nvidia.
Musk has also made it clear that he wants to get paid, a lot. In November, Tesla shareholders will vote on the board’s proposal to pay the CEO a remarkable $1 trillion over the next decade. The deal would also increase Musk’s stake in Tesla from 13 percent to a quarter. But Musk would only get that big figure—and the extra control—if he hits a series of ambitious metrics, including 20 million vehicles delivered, 1 million robotaxis in commercial operation, and an $8.5 trillion valuation. And also, 1 million Optimus humanoid robots delivered.
On a call with investors on Wednesday, Musk locked on to that last point to make his most threatening argument for a gigantic payday yet. “My fundamental concern with regard to how much voting control I have at Tesla is, if I go ahead and build this enormous robot army, can I just be ousted at some point in the future?” he said. “If we build this robot army, do I have at least a strong influence over this robot army? Not control, but a strong influence … I don’t feel comfortable building that robot army unless I have a strong influence.”
Generally, Musk talks about Tesla’s Optimus project as more of a force for peace than war. He’s said that Optimus will upend the job market and free humanity from the drudgery of work. (“Working will be optional, like growing your own vegetables, instead of buying them from the store,” he posted this week.) Elsewhere on the investor call Wednesday, he said that Tesla’s robots would “actually create a world where there is no poverty, where everyone has access to the finest medical care.”
Optimus, he added, “will be an incredible surgeon, and imagine if everyone had access to an incredible surgeon.” For Tesla, Optimus will be “an infinite money glitch,” Musk said, arguing that everyone will want a humanoid robot who can do their work for them.
At Tesla events—and at the Tesla Diner in Los Angeles—Optimus robots are usually seen doing service work: serving drinks and popcorn, or entertaining visitors by dancing or playing rock, paper, scissors. (Optimus participants in a 2024 Tesla event were later acknowledged to be not fully autonomous, but remotely operated by humans.)
Whether Optimus chooses to do laundry or battle, Tesla’s vision of a robotic future still seems a ways away. On Wednesday’s call, Musk dwelled on the challenge of building humanoid hands and forearms, seeming to confirm earlier reporting that the features were proving especially hard for Tesla engineers to hack. And while Tesla set internal goals to produce 5,000 Optimus units this year, The Information reported this month that the company scaled down those production plans over the summer. On Wednesday, Musk said Tesla would have a “production-intent prototype” ready by February or March. Full-scale production, he said, would start at the end of next year.